Projects
Quality Systems Overview
There is such a universe of quality-related topics for pharmaceutical, biologic, and medical device development, it can be quite challenging to sort out, especially for some team members in a fast-moving development project. One goal of this website is to explain quality systems and help put these concepts into perspective. Please see our resources page for links to additional resources for in-depth coverage.
Quality management within a company can be broken out into roughly three major areas: quality assurance (QA), quality control (QC), and current Good Manufacturing Practices (cGMP); Good Laboratory Practices (GLP); and Good Clinical Practices (GCP) that are employed by the manufacturing, laboratory, and clinical staff, respectively. Quality Assurance is chiefly responsible for ensuring quality management through a wide range of tools including batch record review, internal audits, corrective actions, investigations, and training. Quality Control is chiefly responsible for ensuring the accuracy and validity of the analytical data and in some firms is also the group that manages the environmental monitoring (EM) program. The manufacturing, laboratory, and clinical personnel abide by cGMP, GLP, and GCP procedures as they apply to the standard operating procedures (SOPs) of the company.
 When these quality systems are well integrated in a company, the end result is a company culture that quickly identifies manufacturing deviations and/or out-of-specification (OOS) laboratory results and completes thorough investigations in a timely fashion, keeping senior management apprised of the impact to product or the program. The data are trended in a timely fashion and there is evidence of senior management participation in the decision making process as well as being updated on ongoing investigations. Corrective actions are implemented and the impact of those are trended for further action.
When these quality systems are well integrated in a company, the end result is a company culture that quickly identifies manufacturing deviations and/or out-of-specification (OOS) laboratory results and completes thorough investigations in a timely fashion, keeping senior management apprised of the impact to product or the program. The data are trended in a timely fashion and there is evidence of senior management participation in the decision making process as well as being updated on ongoing investigations. Corrective actions are implemented and the impact of those are trended for further action.
A program of ongoing improvement can have immeasurable benefits to the company in terms of client and customer confidence – brand name recognition and association with quality products – as well as better standing with regulatory authorities and inspection teams. Ergo, a company with a solid commitment to quality will enjoy greater efficiency and overall lower cost of goods. And when these quality systems are not well integrated, the impact can be devastating, as seen in product recalls, product seizures, and inspections that complicate movement of materials into different regions of the world. Simply put, once clients leave for another manufacturer or analytical lab, it often takes a dramatic course of events at the firm to convince them to return.
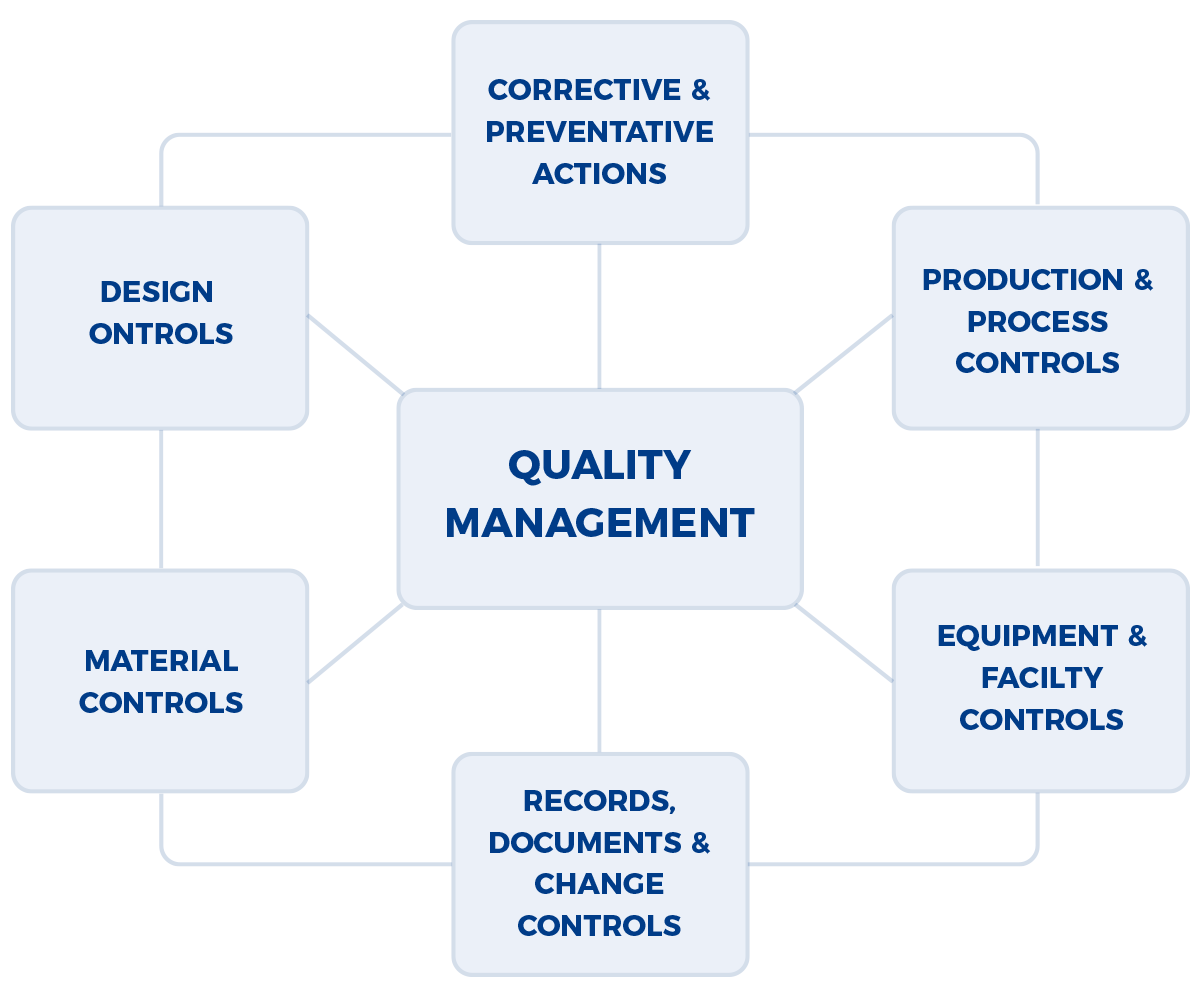 Given the production of pharmaceuticals, biologics, and medical devices is a multi-disciplinary effort (e.g., analytical, manufacturing, building & facilities management, etc.), the FDA (Food & Drug Administration) and other regulatory authorities have developed an audit approach that examines the integrated quality systems used by a company. The approach is well documented in the FDA Quality System Inspection Technique (QSIT) that evaluates how well problems are identified, investigated, reported, trended, and reviewed by senior management. A program that is often set up within companies, a Corrective Action and Preventive Action (CAPA) is framework of integrated standard operating procedures (SOPs) that are meant to address the foundational aspects of QSIT.
Given the production of pharmaceuticals, biologics, and medical devices is a multi-disciplinary effort (e.g., analytical, manufacturing, building & facilities management, etc.), the FDA (Food & Drug Administration) and other regulatory authorities have developed an audit approach that examines the integrated quality systems used by a company. The approach is well documented in the FDA Quality System Inspection Technique (QSIT) that evaluates how well problems are identified, investigated, reported, trended, and reviewed by senior management. A program that is often set up within companies, a Corrective Action and Preventive Action (CAPA) is framework of integrated standard operating procedures (SOPs) that are meant to address the foundational aspects of QSIT.
Essentially, when a manufacturing deviation or out-of-specification (OOS) lab result arises, how well is the event documented and investigated? What kinds of corrective actions were put in place to prevent a re-occurrence? And if an affected lot was released following the investigation, was there sufficient scientific basis to defend the decision? Another important feature is how robust the company’s quality assurance (QA) group is, as demonstrated by the number of internal audits performed or outside vendor audits performed or even how independent they are from senior management when tough decisions are made, such as failing a production lot worth millions of dollars.
These global quality systems evolved largely from the lessons learned from previous failures. The concept of an integrated quality management system is so well established now, they are the central tenets of FDA policy as well as the ICH (International Conference on Harmonization) guidances.
Of the ICH guidance documents, the most relevant to quality systems are detailed in four documents:
Q7: Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients
Q8: Pharmaceutical Development
Q10: Pharmaceutical Quality System
Although the key elements of a global quality system are defined in the guidance documents, over the last decade there has been a shift in how quality is integrated into a product or process. The industry standard has been focused process development and process optimization with varying degrees of connection to the impact on product quality. The result was sometimes a situation where it was not known just how close the validated process parameters were to the ‘edge of failure’ for a product or process. This made justifying manufacturing deviations outside of any validated process extremely difficult to defend, often relying solely on analytical results. Although development reports and development history are now standard components of the Common Technical Dossier, it is more often the case that development data is not written up in a timely fashion, leaving large gaps in the program and resulting in isolated ‘pools of data’ that cannot be bridged.
That is changing with the convergence of Quality by Design (QbD) initiatives that aim to provide a scientific, risk-based holistic and proactive approach to pharmaceutical development. It requires an evaluation of the impact of process parameters on structure-activity relationships. The end result is a development report that provides a rich history of design of engineering (DOE) studies, principal component analysis (PCA), or other statistical measures of the process capability and its impact on product quality parameters. The end result is a more robust process with flexibility for changes.
Globalization
The industry standard is a worldwide manufacturing and distribution network with all the commensurate compliance and regulatory issues for each major region. Creating a harmonized framework for quality assessment and follow up is a rapidly changing and demanding strategic objective. Compliance Strategies, Inc. has worked with several international firms as they harmonized quality programs to support applications and commercial production.
Quality Management Systems: Audits, Training, & Implementation
A large number of pharmaceutical, biologic, and medical device firms contract out production and analytical services. For some companies that are virtual with no in-house quality support, it is vitally important they establish a quality management system for oversight of the manufacturing and analytical services. Compliance Strategies, Inc. and our colleagues have set up quality management systems for numerous firms. Often, the first place to start in the evaluation is an audit followed by an overview of areas for improvement. Once the procedures are written, the staff can be trained and the system brought into compliance.
Customized Quality Support
Many companies only require limited support such as helping write quality procedures, training for certain areas, due diligence audits of vendors, or review of documents and systems. Compliance Strategies, Inc. and their colleagues have enormous experience in the integration of quality assurance and regulatory compliance for every stage of development.
Facility Design & Validation Support
Compliance Strategies, Inc. has worked with several firms for facility upgrade and revalidation, including design of HVAC performance qualification protocols and setting up environmental monitoring programs. The team of experts available is unrivaled in their scope and breadth of experience.
Let's Start a Conversation
Interested in working with us? Call or email.